
thalassemia
Genetics of thalassemia.
Thalassemia is an inherited blood disorder that causes mild or severe anemia. The anemia is due to reduced hemoglobin and fewer red blood cells (erythrocytes) than normal. Hemoglobin is the protein in red blood cells that carries oxygen to all parts of the body.
In people with thalassemia, the genes that code for hemoglobin are missing or variant (different than the normal genes). Severe forms of thalassemia are usually diagnosed in early childhood and are lifelong conditions.
The two main types of thalassemia, alpha and beta, are named for the two protein chains that make up normal hemoglobin. The genes for each type of thalassemia are passed from parents to their children. Alpha and beta thalassemias have both mild and severe forms.
Alpha thalassemia occurs when one or more of the four genes needed for making the alpha globin chain of hemoglobin are variant or missing. Moderate to severe anemia results when more than two genes are affected. The most severe form of alpha thalassemia is known as alpha thalassemia major. It can result in miscarriage.
Beta thalassemia occurs when one or both of the two genes needed for making the beta globin chain of hemoglobin are variant. The severity of illness depends on whether one or both genes are affected and the nature of the abnormality. If both genes are affected, anemia can range from moderate to severe. The severe form of beta thalassemia is also known as Cooley's anemia. Cooley's anemia is the most common severe form of thalassemia in the United States.
Causes
Thalassemia is caused by variant or missing genes that affect how the body makes hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen. People with thalassemia make less hemoglobin and fewer circulating red blood cells than normal. The result is mild or severe anemia.
Many possible combinations of variant genes cause the various types of thalassemia. Thalassemia is always inherited (passed from parents to children). People with moderate to severe forms of thalassemia received variant genes from both parents. A person who inherits a thalassemia gene or genes from one parent and normal genes from the other parent is a carrier (thalassemia trait). Carriers often have no signs of illness other than mild anemia, but they can pass the variant genes on to their children.
Hemoglobin includes two kinds of protein chains called alpha globin chains and beta globin chains. If the problem is with the alpha globin part of hemoglobin, the disorder is alpha thalassemia. If the problem is with the beta globin part, it is called beta thalassemia. There are both mild and severe forms of alpha and beta thalassemia. Severe beta thalassemia is often called Cooley's anemia.
Alpha thalassemia
Four genes are involved in making the alpha globin part of hemoglobin – two from each parent. Alpha thalassemia occurs when one or more of these genes is variant or missing.
If two people with alpha thalassemia trait (carriers) have a child, the baby could have a mild or severe form of alpha thalassemia or could be healthy.
Beta thalassemia
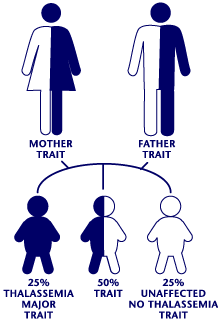
Two genes are involved in making the beta globin part of hemoglobin – one from each parent. Beta thalassemia occurs when one or both of the two genes are variant.
If two people with beta thalassemia trait (carriers) have a baby, one of three things can happen:
Risk factors
Signs and symptoms
The symptoms of thalassemia depend on the type and severity of the disease. Symptoms occur when not enough oxygen gets to various parts of the body due to low hemoglobin and a shortage of red blood cells in the blood (anemia).
"Silent carriers" and persons with alpha thalassemia trait or beta thalassemia trait (also called carriers) usually have no symptoms. Those with alpha or beta thalassemia trait often have mild anemia that may be found by a blood test.
In more severe types of thalassemia, such as Cooley's anemia, signs of the severe anemia are seen in early childhood and may include:
Babies with all four genes affected (a condition called alpha thalassemia major, or hydrops fetalis) usually die before or shortly after birth.
Diagnosis
Thalassemia is diagnosed using blood tests, including a complete blood count (CBC) and special hemoglobin studies.
Cooley's anemia is usually diagnosed in early childhood because of signs and symptoms, including severe anemia. Some people with milder forms of thalassemia may be diagnosed after a routine blood test shows that they have anemia. Doctors suspect thalassemia if a child has anemia and is a member of an ethnic group that is at risk for thalassemia.
To distinguish anemia caused by iron deficiency from anemia caused by thalassemia, tests of the amount of iron in the blood may be done. Iron-deficiency anemia occurs because the body doesn't have enough iron for making hemoglobin. The anemia in thalassemia occurs not because of a lack of iron, but because of a problem with either the alpha globin chain or the beta globin chain of hemoglobin. Iron supplements do nothing to improve the anemia of thalassemia, because missing iron is not the problem.
Family genetic studies are also helpful in diagnosing thalassemia. This involves taking a family history and doing blood tests on family members.
Prenatal testing can determine if an unborn baby has thalassemia and how severe it is likely to be.
Treatment
Treatment for thalassemia depends on the type and severity of the disease.
Blood transfusions
Severe forms of thalassemia are treated by regular blood transfusions. A blood transfusion, given through a needle in a vein, provides blood containing normal red blood cells from healthy donors. In thalassemia treatment, blood transfusions are done on a schedule (often every 2–4 weeks) to keep hemoglobin levels and red blood cell numbers at normal levels. Transfusion therapy can allow a person with severe thalassemia to feel better, enjoy normal activities, and live longer.
Transfusion therapy, while lifesaving, is expensive and carries a risk of transmitting viral and bacterial diseases (for example, hepatitis). Transfusion also leads to excess iron in the blood (iron overload), which can damage the liver, heart, and other parts of the body. To prevent damage, iron chelation therapy is needed to remove excess iron from the body.
Iron chelation therapy
Iron chelation therapy uses medicine to remove the excess iron that builds up in the body when a person has frequent blood transfusions. If the iron is not removed, it damages body organs, such as the heart and liver.
The medicine, deferoxamine, works best when given slowly under the skin, usually with a small portable pump overnight. This therapy is demanding and sometimes is mildly painful, so some people stop chelation therapy. A pill form of iron chelation therapy, deferasirox, was approved in November 2005 for use in the United States.
People who have iron overload should not take vitamins or other supplements that contain iron.
Surgery
Surgery may be needed if body organs, such as the spleen or gall bladder, are affected. For example, if the spleen becomes inflamed and enlarged, it may be removed. If gallstones develop, the gall bladder may be removed.
Bone marrow or stem cell transplants
Bone marrow or stem cell transplants have been used successfully in some children with severe thalassemia. This is a risky procedure, but it offers a cure for those children who qualify.
Other treatments
People with severe thalassemia are more likely to get infections that can worsen their anemia. They should get an annual flu shot and the pneumonia vaccine to help prevent infections.
Folic acid is a B vitamin that helps build red blood cells. People with thalassemia should take folic acid supplements.
Researchers are also studying other treatments, such as gene therapy and fetal hemoglobin.
Gene therapy
Someday, it may be possible to cure thalassemia in an unborn child by inserting a normal gene into the child's stem cells.
Fetal hemoglobin
Researchers are studying ways to enhance production of fetal hemoglobin in people with thalassemia. Fetal hemoglobin is the type of hemoglobin made by the body before birth. After birth, the body usually switches from making fetal hemoglobin to the adult form of hemoglobin. Some children have a gene variant that prevents the switch, and their continuing production of fetal hemoglobin lessens the severity of their illness. Researchers are testing ways to enhance fetal hemoglobin production after birth.
Prevention
Although thalassemia cannot be prevented, it can be identified before birth by prenatal diagnosis.
People who have or believe that they may carry the thalassemia genes can receive genetic counseling to avoid passing the disorder to their children.
Living with thalassemia
The Cooley's Anemia Foundation offers support to people with various types of thalassemia through its Thalassemia Action Group.
If you have moderate or severe thalassemia, you need to take care of your overall health.

