
cystic fibrosis
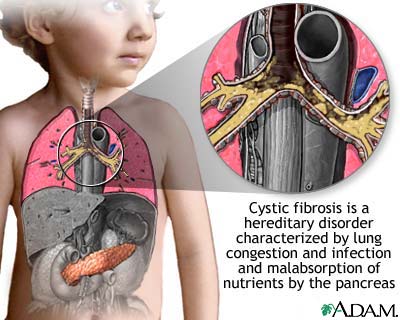
Cystic fibrosis is a hereditary disease the symptoms of which usually appear shortly after birth. These symptoms include faulty digestion, breathing difficulties, and respiratory infections due to mucus accumulation, and excessive loss of salt in sweat. Cystic fibrosis (CF) affects mostly the lungs, pancreas, liver, intestines, sinuses, and sex organs.
Normally, mucus is watery. It keeps the linings of certain organs moist and prevents them from drying out or getting infected. But in CF, an abnormal gene causes mucus to become thick and sticky.
The mucus builds up in the lungs and blocks the airways. This makes it easy for bacteria to grow and leads to repeated serious lung infections. Over time, these infections can cause serious damage to the lungs.
The thick, sticky mucus can also block tubes, or ducts, in the pancreas. As a result, digestive enzymes that are produced by the pancreas cannot reach the small intestine. These enzymes help break down the food that is eaten. Without them, the intestines cannot absorb fats and proteins fully.
As a result:
The abnormal gene also causes the sweat to become extremely salty. As a result, during perspiration, the body loses large amounts of salt. This can upset the balance of minerals in the blood. The imbalance may cause the individual to have a heat emergency.
CF can also cause infertility (mostly in men).
The symptoms and severity of CF vary from person to person. Some people with CF have serious lung and digestive problems. Other people have more mild disease that doesn't show up until they are adolescents or young adults.
Respiratory failure is the most common cause of death in people with CF.
Until the 1980s, most deaths from CF occurred in children and teenagers. Today, with improved treatments, people with CF live, on average, to be more than 35 years old. Research continues to look for better treatments and, ultimately, a cure.
Causes
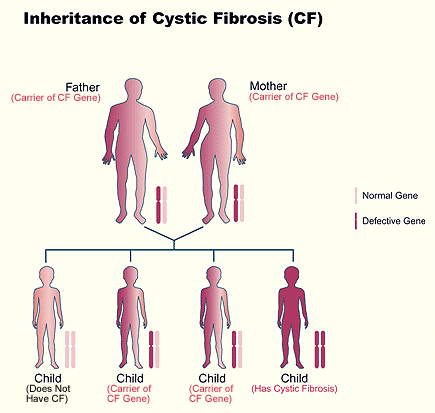
Cystic fibrosis is caused by a defect in a gene called the cystic fibrosis transmembrane conductance regulator (CFTR) gene. This gene makes a protein that controls the movement of salt and water in and out of the cells in the body. In people with CF, the gene does not work effectively. This causes the thick, sticky mucus and very salty sweat that are the main features of CF. Each of us inherits two CFTR genes, one from each parent.
CF carriers:
When two CF carriers have a baby, the baby has a:
Risk factors
About 30,000 people in the United States have cystic fibrosis.
CF is one of the most common inherited diseases among Caucasians.
About 1 in every 3,000 babies born in the United States has CF.
CF is also common in:
CF is much less common among:
About 12 million Americans are carriers of an abnormal CF gene. Many of them do not know that they are CF carriers.
Signs and symptoms
Most of the symptoms of cystic fibrosis are caused by the thick, sticky mucus. The most common symptoms include:
CF can also lead to other medical problems, including:
Diagnosis
First, a doctor will obtain a detailed medical and family history and perform a thorough physical examination. The doctor may order some tests to ensure an accurate diagnosis.
The sweat test is the most useful test for diagnosing cystic fibrosis. It measures the amount of salt in the sweat. For this test, a small amount of a chemical called pilocarpine is rubbed on the patient's arm or leg. An electrode is then attached to this spot. The electrode provides a mild electric current that produces sweat. This may cause tingling or a feeling of warmth. The area is then covered with a gauze pad or filter paper and wrap in plastic. After 30 to 40 minutes, the plastic is removed so the sweat that collected on the pad or paper can be analyzed. The test is usually done twice. High salt levels mean CF.
The following tests may also be carried out to understand more about a patient's condition and how to treat it:
- How much air the lungs can hold
- How quickly the individual can breathe air out of his or her lungs
- How well the lungs add oxygen to and remove carbon dioxide from the blood
- Sputum (phlegm) cultures. A sample of sputum is taken to see what bacteria are growing in it.
If the patient is pregnant, prenatal genetic tests can find out if the baby has CF:
Some States are now testing the blood of all newborns for CF.
CF carrier testing
Individuals may want to check whether they are a CF carrier, if:
A genetics counselor at your local hospital can take a blood or saliva sample to see if it contains the abnormal CFTR gene that causes CF. It will detect 9 out of 10 cases of CF. Some insurance plans cover genetic testing.
Treatment
There still is no cure for cystic fibrosis, but treatments have improved greatly in recent years. The goals of CF treatment are to:
Treatment for lung problems
The main treatments for lung problems in people with CF are:
Antibiotics
Most people with CF have ongoing, low-grade lung infections. Sometimes, these infections become so serious that the individual may need to be hospitalized. Antibiotics are the primary treatment.
Several different types of antibiotics may be given. The choice of antibiotics depends on:
The different types of antibiotics include:
Chest physical therapy
Chest physical therapy (CPT) is also called chest clapping or percussion. It involves pounding the chest and back over and over again to dislodge the mucus from the lungs so that it can be coughed up. CPT for cystic fibrosis should be done three to four times each day.
CPT is also often referred to as postural drainage. This involves the individual sitting or lying on their stomach with their head down while CPT is performed. This allows gravity to help drain the mucus from the lungs.
Because CPT is hard or uncomfortable for some people, several devices have been developed recently that may help with CPT. The devices include:
Several breathing techniques may also help dislodge the mucus. These techniques include:
Exercise
Aerobic exercise helps:
If an individual exercises regularly, he or she may be able to cut back on the chest therapy. A doctor should be consulted before doing this.
Other medications
Anti-inflammatory medications may help reduce the inflammation in the lungs that is caused by ongoing infections. These medications include:
Mucus-thinning drugs reduce the stickiness of mucus in the airways. They include:
Oxygen therapy
If the level of oxygen in the blood is too low, oxygen therapy may be needed. Oxygen is usually given through nasal prongs or a mask.
Lung transplantation
Surgery to replace one or both of the lungs with healthy lungs from a human donor may help the individual. Some of the factors that determine whether a person can undergo lung transplantation include:
Management of digestive problems
Nutritional therapy can improve growth and development, strength, and exercise tolerance. It may also make an individual strong enough to resist some lung infections. Nutritional therapy includes a well-balanced, high-calorie diet that is low in fat and high in protein.
As part of your nutritional therapy, a doctor may:
Other treatments for the digestive problems caused by CF may include:
Living with cystic fibrosis
People with cystic fibrosis should learn as much as they can about the disease and work closely with their doctors to learn how to manage it.
Ongoing medical care is important. Individuals with CF should seek treatment from a team of doctors, nurses, and respiratory therapists who specialize in CF. These specialists are often located at CF Foundation Centers in major medical centers.
Good self-care includes:
People with CF can expect to have a normal sex life.
Having a positive attitude is also helpful.
Parents of someone with CF should not feel guilty about passing it on to their children. And they should not be overprotective; children with CF should be encouraged to be active and self-reliant.

