
phenylketonuria
Phenylketonuria (PKU) is a genetic disorder in which the body cannot metabolize, or process, the essential amino acid phenylalanine (Phe) normally because the enzyme phenylalanine hydroxylase (see below) is absent or inactive. Phe is found in almost all foods. If the Phe level gets too high, it can damage the brain and cause severe mental retardation. All babies born in hospitals in the US and many other countries are now routinely screened for PKU using a heel-prick blood sample. Antenatal screening is also available. This makes it easier to diagnose and treat the problem early.
Children with PKU excrete phenylalanine in their urine. They typically have very fair hair and their urine has a musty smell.
The best treatment for PKU is a diet of low-protein foods. There are special formulas for newborns. For older children and adults, the diet includes many fruits and vegetables. It also includes some low-protein breads, pastas, and cereals. Nutritional formulas provide the vitamins and minerals that children with PKU can't get from their food.
Babies who get on this special diet soon after they are born develop normally. Many have no symptoms of PKU. It is important that they stay on the diet for the rest of their lives.
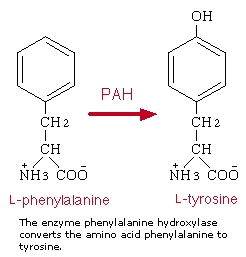
Genetic and biochemical basis of PKU
Classical PKU is an autosomal recessive disorder, caused by mutations in both alleles of the gene for phenylalanine hydroxylase (PAH), found on chromosome 12. In the body, phenylalanine hydroxylase converts the amino acid phenylalanine to tyrosine, another amino acid. Mutations in both copies of the gene for PAH means that the enzyme is inactive or is less efficient, and the concentration of phenylalanine in the body can build up to toxic levels. In some cases, mutations in PAH will result in a phenotypically mild form of PKU called hyperphenylalanemia. Both diseases are the result of a variety of mutations in the PAH locus; in those cases where a patient is heterozygous for two mutations of PAH (ie each copy of the gene has a different mutation), the milder mutation will predominate.

