
acromegaly
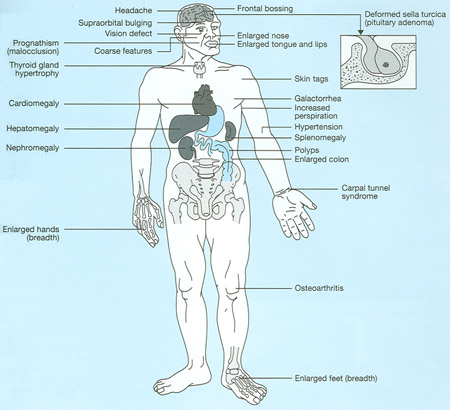
Characteristics of acromegaly.
Acromegaly is a hormonal disorder in which overproduction of growth hormone (GH) by the pituitary gland, commonly after adulthood, causes enlarged hands, feet, and facial features. Usually the excess growth hormone comes from benign, or noncancerous, tumors on the pituitary. These benign tumors are called adenomas.
Acromegaly is most often diagnosed in middle-aged adults, although symptoms can appear at any age. If not treated, acromegaly can result in serious illness and premature death. Acromegaly is treatable in most patients, but because of its slow and often "sneaky" onset, it often is not diagnosed early or correctly. The most serious health consequences of acromegaly are type 2 diabetes mellitus, high blood pressure, increased risk of cardiovascular disease, and arthritis. Patients with acromegaly are also at increased risk for colon polyps, which may develop into colon cancer if not removed.
When GH-producing tumors occur in childhood, the disease that results is called gigantism rather than acromegaly. A child's height is determined by the length of the so-called long bones in the legs. In response to GH, these bones grow in length at the growth plates – areas near either end of the bone. Growth plates fuse after puberty, so the excessive GH production in adults does not result in increased height. However, prolonged exposure to excess GH before the growth plates fuse causes increased growth of the long bones and thus increased height. Pediatricians may become concerned about this possibility if a child's growth rate suddenly and markedly increases beyond what would be predicted by previous growth and how tall the child's parents are.
Symptoms
The name acromegaly comes from the Greek words for "extremities" and "enlargement," reflecting one of its most common symptoms – the abnormal growth of the hands and feet. Swelling of the hands and feet is often an early feature, with patients noticing a change in ring or shoe size, particularly shoe width. Gradually, bone changes alter the patient's facial features: The brow and lower jaw protrude, the nasal bone enlarges, and the teeth space out.
Overgrowth of bone and cartilage often leads to arthritis. When tissue thickens, it may trap nerves, causing carpal tunnel syndrome, which results in numbness and weakness of the hands. Body organs, including the heart, may enlarge.
Other symptoms of acromegaly include:
Cause
Acromegaly is caused by prolonged overproduction of GH by the pituitary gland. The pituitary produces several important hormones that control body functions such as growth and development, reproduction, and metabolism. But hormones never seem to act simply and directly. They usually "cascade" or flow in a series, affecting each other's production or release into the bloodstream.
GH is part of a cascade of hormones that, as the name implies, regulates the physical growth of the body. This cascade begins in a part of the brain called the hypothalamus. The hypothalamus makes hormones that regulate the pituitary. One of the hormones in the GH series, or "axis," is growth hormone-releasing hormone (GHRH), which stimulates the pituitary gland to produce GH.
Secretion of GH by the pituitary into the bloodstream stimulates the liver to produce another hormone called insulin-like growth factor I (IGF-I). IGF-I is what actually causes tissue growth in the body. High levels of IGF-I, in turn, signal the pituitary to reduce GH production.
The hypothalamus makes another hormone called somatostatin, which inhibits GH production and release. Normally, GHRH, somatostatin, GH, and IGF-I levels in the body are tightly regulated by each other and by sleep, exercise, stress, food intake, and blood sugar levels. If the pituitary continues to make GH independent of the normal regulatory mechanisms, the level of IGF-I continues to rise, leading to bone overgrowth and organ enlargement. High levels of IGF-I also cause changes in glucose (sugar) and lipid (fat) metabolism and can lead to diabetes, high blood pressure, and heart disease.
Pituitary tumors
In more than 95% of people with acromegaly, a benign tumor of the pituitary gland, called an adenoma, produces excess GH. Pituitary tumors are labeled either micro- or macro-adenomas, depending on their size. Most GH-secreting tumors are macro-adenomas, meaning they are larger than 1 centimeter. Depending on their location, these larger tumors may compress surrounding brain structures. For example, a tumor growing upward may affect the optic chiasm – where the optic nerves cross – leading to visual problems and vision loss. If the tumor grows to the side, it may enter an area of the brain called the cavernous sinus where there are many nerves, potentially damaging them.
Compression of the surrounding normal pituitary tissue can alter production of other hormones. These hormonal shifts can lead to changes in menstruation and breast discharge in women and erectile dysfunction in men. If the tumor affects the part of the pituitary that controls the thyroid – another hormone-producing gland – then thyroid hormones may decrease. Too little thyroid hormone can cause weight gain, fatigue, and hair and skin changes. If the tumor affects the part of the pituitary that controls the adrenal gland, the hormone cortisol may decrease. Too little cortisol can cause weight loss, dizziness, fatigue, hypotension (low blood pressure), and nausea.
Some GH-secreting tumors may also secrete too much of other pituitary hormones. For example, they may produce prolactin, the hormone that stimulates the mammary glands to produce milk. Rarely, adenomas may produce thyroid-stimulating hormone. Doctors should assess all pituitary hormones in people with acromegaly.
Rates of GH production and the aggressiveness of the tumor vary greatly among people with adenomas. Some adenomas grow slowly and symptoms of GH excess are often not noticed for many years. Other adenomas grow more rapidly and invade surrounding brain areas or the venous sinuses, which are located near the pituitary gland. Younger patients tend to have more aggressive tumors. Regardless of size, these tumors are always benign.
Most pituitary tumors develop spontaneously and are not genetically inherited. They are the result of a genetic alteration in a single pituitary cell, which leads to increased cell division and tumor formation. This genetic change, or mutation, is not present at birth, but happens later in life. The mutation occurs in a gene that regulates the transmission of chemical signals within pituitary cells. It permanently switches on the signal that tells the cell to divide and secrete GH. The events within the cell that cause disordered pituitary cell growth and GH oversecretion currently are the subject of intensive research.
Nonpituitary tumors
Rarely, acromegaly is caused not by pituitary tumors but by tumors of the pancreas, lungs, and other parts of the brain. These tumors also lead to excess GH, either because they produce GH themselves or, more frequently, because they produce GHRH, the hormone that stimulates the pituitary to make GH. When these non-pituitary tumors are surgically removed, GH levels fall and the symptoms of acromegaly improve.
In patients with GHRH-producing, non-pituitary tumors, the pituitary still may be enlarged and may be mistaken for a tumor. Physicians should carefully analyze all "pituitary tumors" removed from patients with acromegaly so they do not overlook the rare possibility that a tumor elsewhere in the body is causing the disorder.
How common is acromegaly?
Small pituitary adenomas are common, affecting about 17% of the population. However, research suggests most of these tumors do not cause symptoms and rarely produce excess GH. Scientists estimate that three to four out of every million people develop acromegaly each year and about 60 out of every million people suffer from the disease at any time. Because the clinical diagnosis of acromegaly is often missed, these numbers probably underestimate the frequency of the disease.
Diagnosis
Blood tests
If acromegaly is suspected, a doctor must measure the GH level in a person's blood to determine if it is elevated. However, a single measurement of an elevated blood GH level is not enough to diagnose acromegaly: Because GH is secreted by the pituitary in impulses, or spurts, its concentration in the blood can vary widely from minute to minute. At a given moment, a person with acromegaly may have a normal GH level, whereas a GH level in a healthy person may even be five times higher.
More accurate information is obtained when GH is measured under conditions that normally suppress GH secretion. Health care professionals often use the oral glucose tolerance test to diagnose acromegaly because drinking 75 to 100 grams of glucose solution lowers blood GH levels to less than 1 nanogram per milliliter (ng/ml) in healthy people. In people with GH overproduction, this suppression does not occur. The oral glucose tolerance test is a highly reliable method for confirming a diagnosis of acromegaly.
Physicians also can measure IGF-I levels, which increase as GH levels go up, in people with suspected acromegaly. Because IGF-I levels are much more stable than GH levels over the course of the day, they are often a more practical and reliable screening measure. Elevated IGF-I levels almost always indicate acromegaly. However, a pregnant woman's IGF-I levels are two to three times higher than normal. In addition, physicians must be aware that IGF-I levels decline with age and may also be abnormally low in people with poorly controlled diabetes or liver or kidney disease.
Imaging
After acromegaly has been diagnosed by measuring GH or IGF-I levels, a magnetic resonance imaging (MRI) scan of the pituitary is used to locate and detect the size of the tumor causing GH overproduction. MRI is the most sensitive imaging technique, but computerized tomography (CT) scans can be used if the patient should not have MRI. For example, people who have pacemakers or other types of implants containing metal should not have an MRI scan because MRI machines contain powerful magnets.
If a head scan fails to detect a pituitary tumor, the physician should look for non-pituitary "ectopic" tumors in the chest, abdomen, or pelvis as the cause of excess GH. The presence of such tumors usually can be diagnosed by measuring GHRH in the blood and by a CT scan of possible tumor sites.
Rarely, a pituitary tumor secreting GH may be too tiny to detect even with a sensitive MRI scan.
Treatment
Currently, treatment options include surgical removal of the tumor, medical therapy, and radiation therapy of the pituitary.
Goals of treatment are to:
Surgery
Surgery is the first option recommended for most people with acromegaly, as it is often a rapid and effective treatment. The surgeon reaches the pituitary via an incision through the nose or inside the upper lip and, with special tools, removes the tumor tissue in a procedure called transsphenoidal surgery. This procedure promptly relieves the pressure on the surrounding brain regions and leads to a rapid lowering of GH levels. If the surgery is successful, facial appearance and soft tissue swelling improve within a few days.
Surgery is most successful in patients with blood GH levels below 45 ng/ml before the operation and with pituitary tumors no larger than 10 millimeters (mm) in diameter. Success depends in large part on the skill and experience of the surgeon, as well as the location of the tumor. Even with the most experienced neurosurgeon, the chance of a cure is small if the tumor has extended into critical brain structures or into the cavernous sinus where surgery could be risky.
The success rate also depends on what level of GH is defined as a cure. The best measure of surgical success is normalization of GH and IGF-I levels. The overall rate of remission – control of the disease – after surgery ranges from 55 to 80%.
A possible complication of surgery is damage to the surrounding normal pituitary tissue, which requires lifelong use of pituitary hormone replacement. The part of the pituitary that stores antidiuretic hormone – a hormone important in water balance – may be temporarily or, rarely, permanently damaged and the patient may require medical therapy. Other potential problems include cerebrospinal fluid leaks and, rarely, meningitis. Cerebrospinal fluid bathes the brain and can leak from the nose if the incision area doesn't heal well. Meningitis is a bacterial or viral infection of the meninges, the outer covering of the brain.
Even when surgery is successful and hormone levels return to normal, people with acromegaly must be carefully monitored for years for possible recurrence of the disease. More commonly, hormone levels improve, but do not return to normal. Additional treatment, usually medications, may be required.
Medical therapy
Medical therapy is most often used if surgery does not result in a cure and sometimes to shrink large tumors before surgery. Three medication groups are used to treat acromegaly.
Somatostatin analogs (SSAs) are the first medication group used to treat acromegaly. They shut off GH production and are effective in lowering GH and IGF-I levels in 50 to 70% of patients. SSAs also reduce tumor size in around 0 to 50% of patients but only to a modest degree. Several studies have shown that SSAs are safe and effective for long-term treatment and in treating patients with acromegaly caused by nonpituitary tumors. Long-acting SSAs are given by intramuscular injection once a month.
Digestive problems – such as loose stools, nausea, and gas – are a side effect in about half of people taking SSAs. However, the effects are usually temporary and rarely severe. About 10 to 20% of patients develop gallstones, but the gallstones do not usually cause symptoms. In rare cases, treatment can result in elevated blood glucose levels. More commonly, SSAs reduce the need for insulin and improve blood glucose control in some people with acromegaly who already have diabetes.
The second medication group is the GH receptor antagonists (GHRAs), which interfere with the action of GH. They normalize IGF-I levels in more than 90 percent of patients. They do not, however, lower GH levels. Given once a day through injection, GHRAs are usually well-tolerated by patients. The long-term effects of these drugs on tumor growth are still under study. Side effects can include headaches, fatigue, and abnormal liver function.
Dopamine agonists make up the third medication group. These drugs are not as effective as the other medications at lowering GH or IGF-I levels, and they normalize IGF-I levels in only a minority of patients. Dopamine agonists are sometimes effective in patients who have mild degrees of excess GH and have both acromegaly and hyperprolactinemia – too much of the hormone prolactin. Dopamine agonists can be used in combination with SSAs. Side effects can include nausea, headache, and lightheadedness.
Radiation therapy
Radiation therapy is usually reserved for people who have some tumor remaining after surgery and do not respond to medications. Because radiation leads to a slow lowering of GH and IGF-I levels, these patients often also receive medication to lower hormone levels. The full effect of this therapy may not occur for many years.
The two types of radiation delivery are conventional and stereotactic. Conventional radiation delivery targets the tumor with external beams but can damage surrounding tissue. The treatment delivers small doses of radiation multiple times over 4 to 6 weeks, giving normal tissue time to heal between treatments.
Stereotactic delivery allows precise targeting of a high-dose beam of radiation at the tumor from varying angles. The patient must wear a rigid head frame to keep the head still. The types of stereotactic radiation delivery currently available are proton beam, linear accelerator (LINAC), and gamma knife. With stereotactic delivery, the tumor must be at least 5 mm from the optic chiasm to prevent radiation damage. This treatment can sometimes be done in a single session, reducing the risk of damage to surrounding tissue.
All forms of radiation therapy cause a gradual decline in production of other pituitary hormones over time, resulting in the need for hormone replacement in most patients. Radiation also can impair a patient's fertility. Vision loss and brain injury are rare complications. Rarely, secondary tumors can develop many years later in areas that were in the path of the radiation beam.
Which treatment for acromegaly is most effective?
No single treatment is effective for all patients. Treatment should be individualized, and often combined, depending on patient characteristics such as age and tumor size.
If the tumor has not yet invaded surrounding nonpituitary tissues, removal of the pituitary adenoma by an experienced neurosurgeon is usually the first choice. Even if a cure is not possible, surgery may be performed if the patient has symptoms of neurological problems such as loss of peripheral vision or cranial nerve problems. After surgery, hormone levels are measured to determine whether a cure has been achieved. This determination can take up to 8 weeks because IGF-I lasts a long time in the body's circulation. If cured, a patient must be monitored for a long time for increasing GH levels.
If surgery does not normalize hormone levels or a relapse occurs, an endocrinologist should recommend additional drug therapy. With each medication, long-term therapy is necessary because their withdrawal can lead to rising GH levels and tumor re-expansion.
Radiation therapy is generally reserved for patients whose tumors are not completely removed by surgery, who are not good candidates for surgery because of other health problems, or who do not respond adequately to surgery and medication.


