
Marfan syndrome
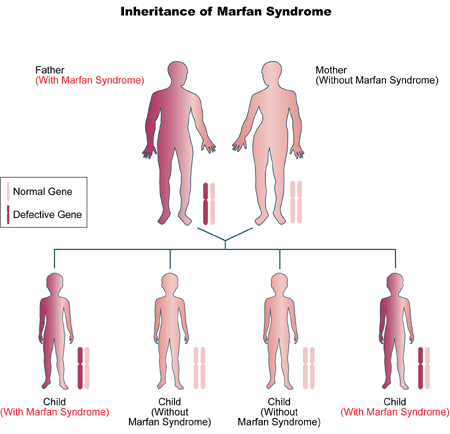
Figure 1. Inheritance of Marfan syndrome.
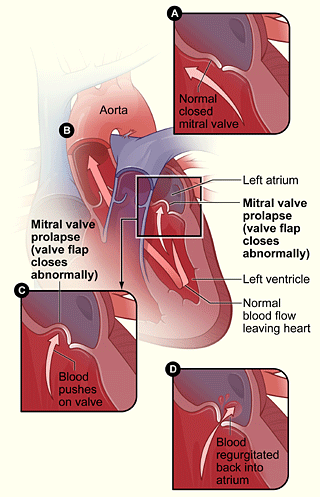
Figure 2. (A) shows the normal mitral valve separating the left atrium from the left ventricle. (B) shows the heart with mitral valve prolapse. (C) shows the detail of mitral valve prolapse. (D) shows a mitral valve that allows blood to flow backward into the left atrium.
Marfan syndrome is a rare, inherited disorder of connective tissue – tissue holds all the parts of the body together and helps control growth. Connective tissue gets some of its strength from a protein called fibrillin 1, which also plays an important role in controlling the growth and development of the body. In Marfan syndrome, the body produces fibrillin 1 that does not work properly. As a result, the connective tissue is not as strong as it should be, there are abnormalities of the skeleton, heart, and eyes, and the growth and development of the body are affected. Marfan syndrome affects about 2 in every 100,000 people. See also connective tissue disorders.
Effects of Marfan syndrome on the body
Heart and blood vessels
Most people who have Marfan syndrome have problems with their heart and blood vessels because of weak connective tissue.
Heart valves may not close properly and may let blood leak backward. The aorta – the large artery that carries blood away from the heart out to the body – may stretch, enlarge, and even burst. The aorta may also develop a tear in its inner wall. This tear can expand and block blood flow through the aorta. This is called aortic dissection.
Bones, cartilage, and ligaments
People with Marfan syndrome tend to have tall, slender bodies with arms and legs disproportionately long compared to the trunk. They also usually have long fingers and toes. The ligaments and joints are typically loose. Because of rib overgrowth, the chest may protrude or be indented. Abnormal curving of the spine, called scoliosis, lordosis, or kyphosis, can occur.
Eyes
The most common vision problem in people with Marfan syndrome is myopia (near-sightedness). Myopia is sharp vision for things that are close and blurred vision for things that are farther away. Dislocation of the lens of the eye is a hallmark of Marfan syndrome that occurs in very few other conditions. Cataracts, or glaucoma at an early age, or detached retinas, also occur in people with Marfan syndrome.
Lungs
In Marfan syndrome, the air sacs (alveoli) in the lungs may be unusually large. In addition, the chest may be abnormally shaped. For these reasons, people with Marfan syndrome may have breathing problems, such as collapsed lungs (spontaneous pneumothorax) and early emphysema.
Skin
Stretch marks, not due to weight gain or loss, commonly occur on the skin of people with Marfan syndrome.
Outlook
Marfan syndrome is a lifelong disorder with no known cure. As recently as the 1970s, most people with Marfan syndrome did not survive beyond age 40–50. Some infants and children died from complications due to the condition. In most cases, death was due to heart and blood vessel problems.
Today, because of early diagnosis and better medical and surgical treatments, people with Marfan syndrome can live longer and healthier lives, with fewer and less severe complications. However, aging with Marfan syndrome poses new concerns as this chronic and progressive disorder advances. In addition, those who are not diagnosed or treated are still at risk of an early sudden death due to blood vessel problems.
Cause
Marfan syndrome is caused by a defect in the gene that controls the structure of a protein called fibrillin 1. Fibrillin 1 is an important part of connective tissue. If you have the defective gene, your body produces fibrillin 1 that does not function as it should. As a result, your connective tissue is not as strong as it should be, and it weakens over time. Fibrillin 1 also plays a role in the growth and development of many of the organs of the body. Defective fibrillin 1 contributes to the tall, thin body type seen in people with Marfan syndrome, as well as other Marfan signs and symptoms.
About 3 out of 4 people with Marfan syndrome inherit the defective gene from a parent. In about 1 out of every 4 people with Marfan syndrome, the gene abnormality occurs due to a chance (spontaneous) mutation of the gene.
Who is at risk?
Marfan syndrome is a hereditary disorder passed from parent to child (Figure 1). Every person has two copies of every gene in the body (except some genes related to gender). One copy of each gene pair is inherited from each parent. It only takes one copy of the defective gene, inherited from one parent, for a person to have Marfan syndrome.
If one parent of a couple has Marfan syndrome, each of their children has a 50 percent chance of inheriting the Marfan gene.
If both parents have Marfan syndrome, each of their children has a 1 out of 4 chance of inheriting two normal copies of the gene, a 2 out of 4 chance of inheriting one copy of the Marfan gene, and a 1 out of 4 chance of inheriting two copies of the Marfan gene. Pregnancies in which the fetus inherits two copies of the Marfan gene are rare. Infants born with two copies of the Marfan gene are severely affected and usually do not survive beyond infancy.
Signs and symptoms
The signs and symptoms of Marfan syndrome vary from one person to another, even within the same family. Some people have mild signs and symptoms, while others may have severe problems and discomfort. Signs and symptoms occur in many parts of the body, including:
Appearance and body build
Some of the major signs of Marfan syndrome are the common physical features seen in people with the condition. People with Marfan syndrome often have:
Bones, cartilage, and ligaments
The bones of the limbs, hands, and feet often grow too long in people with Marfan syndrome. This typically leads to a tall, thin body with disproportionately long arms, fingers, legs, and toes. People with Marfan syndrome have loose, relaxed ligaments and are usually loose jointed.
Chest abnormalities may occur due to an overgrowth of the ribs. There are two types of chest abnormalities:
Curvature of the spine may occur. It usually develops during childhood, often gets worse during the teenage growth spurt, and may require surgical treatment. The three main types of abnormal spine curvature are:
Curvature of the spine can cause back pain, posture problems, and deformity. Scoliosis can sometimes reduce lung function.
Another problem that can occur is dural ectasia, which is the stretching of the membrane surrounding the brain and spinal cord. Dural ectasia can wear down the bones of the spine over time. Symptoms may include:
Dural ectasia is a hallmark of Marfan syndrome that is very rare in the general population.
Heart and blood vessels
People who have Marfan syndrome often have problems with the heart and blood vessels. The valves of the heart may not work properly and may permit some blood flow to be reversed, causing the heart to do extra work. The aorta – the large artery that carries blood away from the heart to the body – may stretch and enlarge. These problems can cause signs and symptoms, such as:
Two of the heart and blood vessel complications that can occur in people with Marfan syndrome are mitral valve prolapse (MVP) and enlargement of the aorta.
Mitral valve prolapse
MVP (Figure 2) is a problem with the heart's mitral valve. The mitral valve controls the flow of blood through two of the chambers in the heart, the left atrium and the left ventricle. The flaps of the valve are designed to allow blood to flow in one direction – from the left atrium into the left ventricle – and to prevent blood from flowing the other way.
In MVP, the mitral valve does not work correctly. The valve flaps are large
and floppy. They may overlap or not close completely. One or both flaps
may flutter or swing back into the atrium.
The abnormal mitral valve can allow blood to flow backward through the valve
in the wrong direction. This is called mitral
regurgitation. When this happens, the heart has to work harder to make
up for the backward flow of blood. Over time, the heart can become overworked,
leading to heart failure.
MVP occurs in about 3 out of every 4 people with Marfan syndrome. It also occurs in people who do not have Marfan syndrome. In many people with MVP, little or no blood leaks back through the valve, there are no symptoms, and no treatment is needed. In some people, blood does leak back through the valve, and these people may have symptoms and require treatment.
Enlargement of the aorta (Aortic dilation)
The aorta is the large artery that carries oxygen-rich blood away from your heart out to the rest of the body. When weak connective tissue causes the walls of the aorta to weaken, the aorta widens and stretches. Most often, the enlargement begins where the aorta connects with the left ventricle of the heart, just above the aortic valve. This part of the aorta is called the aortic root.
Enlargement of the aorta can lead to several serious complications:
Eyes and vision
Nearsightedness (myopia) is the most common eye problem in people with Marfan syndrome. It can range from mild to severe. People with Marfan syndrome often have astigmatism, which causes visual distortion and blurred vision.
Dislocation of the lens of the eye (ectopia lentis) is another common complication of Marfan syndrome. It is considered a hallmark of Marfan syndrome because it occurs in very few other conditions. It may affect one or both eyes, and the lens may be higher or lower than normal or shifted off to the side. Vision in the affected eye(s) may be severely affected.
Other, less common eye problems in Marfan syndrome include detached retina, cataracts, and glaucoma. A detached retina occurs when there are holes or tears in the inner lining of the eye. A cataract is a clouding of the lens. Glaucoma occurs as a result of high pressure in the eye. People with Marfan syndrome tend to get cataracts and glaucoma at a younger age than people who do not have Marfan syndrome.
Lungs
In Marfan syndrome, the air sacs in the lungs may be larger than normal. This can cause breathing problems. People with Marfan syndrome are at an increased risk of developing emphysema or chronic obstructive pulmonary disease (COPD), a serious lung disease.
Sudden collapse of the lung, called spontaneous pneumothorax can occur in people with Marfan syndrome. About 1 in every 20 people with Marfan syndrome develops this problem. Collapsed lung can happen after only a slight blow to the chest, or even while at rest without a known cause. Collapsed lung usually causes sudden shortness of breath and requires immediate medical attention.
Sleep apnea is a problem that is often associated in the general population with being overweight or obese. But thin people with Marfan syndrome can also suffer from it. This may be due to looseness of the connective tissue in the airways.
Skin
Stretch marks on the skin occur in about 2 out of every 3 people with Marfan syndrome. The stretch marks are usually on the lower back, buttocks, shoulders, breasts, thighs, and abdomen. They differ from stretch marks in the general population because, in people with Marfan syndrome, they occur without excessive weight gain or loss. The marks usually appear around the time of puberty, but may occur in childhood. They do not require treatment.
Marfan syndrome is diagnosed based on three main factors:
Diagnostic criteria
The diagnosis of Marfan syndrome is based on a person's family history and the presence of certain findings on physical exam. Lab and genetic tests can help, but by themselves are not enough to diagnose Marfan syndrome.
Doctors use a set of guidelines called the Ghent criteria to establish the diagnosis of Marfan syndrome. The Ghent criteria specify combinations of family history and physical findings that need to be present in order to diagnose Marfan syndrome. The criteria were updated in 1996 and named for the city in which the expert panel that decided the criteria met. The previous guidelines were called the Berlin criteria.
It is important to note that a person may have some of the features of Marfan syndrome, but not enough to actually be diagnosed with the disorder. In this situation, it is important for the person to be treated for the signs and symptoms he or she does have.
In some cases, Marfan syndrome may not be diagnosed until early adulthood, when the signs and symptoms become more obvious. Infants and children may be diagnosed if the signs and symptoms are obvious or if they are being treated for some other condition. Sometimes Marfan syndrome is identified in children or young adults after a family member has been diagnosed.
Specialists involved
A family doctor or pediatrician may observe certain signs and symptoms that suggest Marfan syndrome and refer a person to a specialist for diagnosis. Because Marfan syndrome affects so many body systems, several specialists may be involved, including:
Diagnostic tests
Doctors may use a combination of tests to diagnose Marfan syndrome. In addition, some tests also may be used for monitoring patients and detecting complications. Tests may include:
There are several different types of echocardiograms, including a stress echocardiogram. During this test, an echocardiogram is done both before and after your heart is stressed either by having you exercise or by injecting a medicine into your bloodstream that makes your heart beat faster and work harder. A stress echocardiogram is usually done to find out if you have decreased blood flow to your heart (coronary artery disease).
Treatment
Treatments are available for Marfan syndrome to limit and prevent complications, prevent death from heart-related complications, improve physical appearance, and limit disfigurement. There is new hope for treatments that will be directed at how fibrillin 1 controls the growth in the body. Currently, Marfan syndrome has no cure.
Treatment for heart and blood vessel problems
Treatment for problems with the heart and blood vessels may include medicines and surgery. To decide when treatment is needed, doctors should repeat diagnostic tests (such as an annual echocardiogram) periodically and look for changes in the heart and aorta. Lifestyle changes also can help to reduce strain and stress on the heart and blood vessels.
Medicines
A variety of medicines are used to treat heart and blood vessel problems in people with Marfan syndrome.
Beta blockers
Beta blockers are often used to:
Calcium channel blockers
Calcium channel blockers:
Angiotensin-converting enzyme inhibitors
Also called ACE inhibitors, these medicines:
Other medicines may include:
Surgery
Aortic valve surgery
Some people with Marfan syndrome need surgery to repair or replace the aorta or the aortic valve. Doctors may recommend surgery for several reasons, such as:
Although any kind of surgery has risks, doctors have had more success with aortic repair and aortic valve replacement surgery when done on an elective (non-emergency) basis than on an emergency basis.
Patients with the criteria mentioned above may need surgery to repair or replace their aorta. Composite valve-graft aortic replacement surgery and aortic valve-sparing surgery are the two main types of surgery that surgeons use in patients with Marfan syndrome and problems with the aorta. Choices for aortic repair or replacement surgery each have advantages and disadvantages. These should be thoroughly discussed with the surgeon to determine which surgery is best suited to the individual.
Composite valve-graft aortic replacement surgery.
Most often, doctors recommend composite valve-graft surgery to repair an enlarged aorta or to prevent aortic dissection and rupture. In this open-heart surgery, the aortic valve and part of the aorta are replaced. Surgeons remove the aneurysm (enlarged part of the aorta) and replace it with an artificial tube called a graft. An artificial (mechanical) valve replaces the native aortic valve. People who receive a mechanical aortic valve require anticoagulant medicine to prevent blood clots from forming on the mechanical valve.
Aortic valve-sparing surgery.
Valve-sparing surgery is a newer and increasingly used approach for patients who have an enlarged aorta but an otherwise normal aortic valve. Similar to composite valve-graft surgery, valve-sparing surgery is an open-heart procedure. Most surgeons do not recommend valve-sparing surgery in emergency situations.
During valve-sparing surgery, the enlarged part of the aorta is removed. A tube, or graft, is tailored appropriately, and the patient's own, healthy aortic valve is sewn within the tube.
When compared to composite valve-graft surgery, advantages of this type of surgery include:
Mitral valve surgery
Surgery may be needed if a person with Marfan syndrome has mitral valve prolapse or if the valve is leaking (mitral regurgitation). The doctor may suggest valve repair procedures (valvuloplasty) or valve replacement.
Treatment for skeletal system problems
Doctors may recommend several options to treat problems of the skeletal system.
Brace or other prosthetic device
These devices may be used to stabilize the spine. They are often used in children with scoliosis if the curve is between 20 and 40 degrees. A brace helps keep the curvature from getting worse as a child grows, but surgery may still be necessary.
Surgery for scoliosis
If the curve in the spine is greater than 40 to 50 degrees, surgery may be needed. Doctors may suggest surgery if a person has severe, ongoing back pain or if lung function is being affected. In this surgery, the surgeon inserts metal rods that help straighten and fuse the spine in the correct position.
Surgery for chest wall (pectus) deformities
Sometimes people with Marfan syndrome need chest surgery to limit damage and disfigurement, to prevent heart and lung compression or impaired function, or to improve their appearance. Doctors usually wait to do this surgery until after mid-adolescence when the ribs stop growing. Surgery to treat "pigeon breast" flattens and straightens the deformed breastbone and ribs. Surgery to treat "funnel chest" raises and straightens the breastbone and ribs.
Treatment for eye problems
Several eye conditions that often occur in people with Marfan syndrome need treatment. These include nearsightedness (myopia), visual distortion (astigmatism), and dislocated lens (ectopia lentis).
The doctor may recommend:
Living with Marfan syndrome
Early diagnosis and advances in medical care have greatly improved the quality and length of life for people with Marfan syndrome. Finding and treating problems early can prevent or delay complications and improve quality of life.
Ongoing health care needs
If you have Marfan syndrome, you should have regular checkups with specialists. These checkups include:
If you have Marfan syndrome, your doctor may suggest that you:
Emotional and psychological needs
Having Marfan syndrome can be emotionally stressful. You and your family may find it difficult to accept and adjust to the disorder. Fortunately, information and support are available.
Your doctors can give you accurate information about Marfan syndrome and how to take care of it. They can answer your questions about the disorder and its treatment. The National Marfan Foundation also offers extensive information.
Social support may be available from family members and friends, other people with Marfan syndrome and their family members, and support groups. Peer counseling and support are available through the National Marfan Foundation.
Genetic counseling or other professional counseling (for example, from a social worker or psychologist) may be useful.
Physical activity
Your doctor may recommend gentle, regular exercise. An exercise program can be tailored to your needs. In general, low-impact exercise can help improve physical strength and endurance, increase bone density, and lower blood pressure. But more strenuous activities may be dangerous, increasing the risk of damage to your aorta, eyes, and ligaments. The doctor can advise you about safe types of exercise, as well as activities to avoid, such as contact sports and competitive school sports.
People with Marfan syndrome often are tall and loose jointed. Young people with this type of body build may be encouraged to play basketball or other sports before they find out that they have Marfan syndrome. Testing of young people with this kind of body build – or with a family history of Marfan syndrome – may lead to early diagnosis and prevent complications.
Pregnancy in women with Marfan syndrome
Having Marfan syndrome increases the risk of serious complications during pregnancy because of the added stress on the heart. However, many pregnant women with Marfan syndrome have safe, normal deliveries.
If you are a woman with Marfan syndrome thinking about having a baby, talk to your doctor about the risks to you and your baby. For example:
Couples considering pregnancy may want to seek genetic counseling if either person has Marfan syndrome.
If you become pregnant, you may want to seek care from a specialist familiar with Marfan syndrome or an obstetrician who specializes in high-risk pregnancies if a Marfan specialist is not available. You should have an echocardiogram every 6–10 weeks throughout pregnancy. Your doctor may recommend bed rest if aortic enlargement or aortic valve leakage occurs.
Anticoagulant medicines such as heparin and Coumadin (warfarin) are often used to reduce the risk of blood clots in women with Marfan syndrome. If you are taking blood-thinning medications and become pregnant, or are planning on becoming pregnant, your doctor may want to change your medicines. Coumadin (warfarin), for example, has been associated with birth defects when taken during the first trimester.
Your doctor may prescribe beta blockers if you have moderate or severe aortic dilation. Beta blockers do not increase the risk of birth defects or harm your unborn baby.

