
sickle cell anemia

The sickle-shaped red blood cells tend to get stuck in blood vessels, blocking the flow of blood.
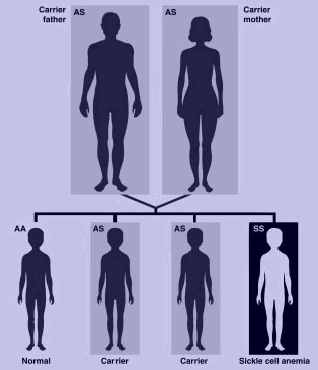
The presence of two sickle cell genes (SS) is needed for sickle cell anemia. If each parent carries one sickle hemoglobin gene (S) and one normal hemoglobin gene (A), with each pregnancy, there is a 25% chance of the child's inheriting two SS genes and having sickle cell anemia; a 25% chance of inheriting two AA genes and not having the disease or being a carrier; and a 50% chance of being an unaffected carrier (AS) like the parents.
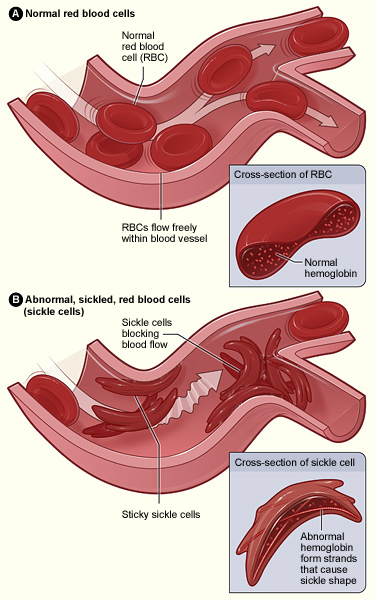
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. Figure B shows abnormal, sickled red blood cells clumping and blocking blood flow in a blood vessel. (Other cells also may play a role in this clumping process.) The inset image shows a cross-section of a sickle cell with abnormal hemoglobin.
Sickle cell anemia is a serious disease in which the body makes abnormally shaped red blood cells. Normal red blood cells are smooth and round like a doughnut without a hole. They move easily through blood vessels to carry oxygen to all parts of the body. In sickle cell anemia, the body produces red blood cells that are shaped like a sickle (or crescent). These "sickle cells" are hard and sticky and they don't move easily through blood vessels. They tend to get stuck and block the flow of blood to the limbs and organs. This can cause pain, organ damage, and a low blood count (anemia).
Sickle cell anemia is an inherited (genetic) disorder seen most commonly in people of African ancestry. The disorder is caused by a single base pair change in one of the genes that codes for hemoglobin, the blood protein that carries oxygen. People who have sickle cell anemia are born with it. It is a lifelong disease.
Sickle cell anemia affects millions of people. Effective treatments exist for the symptoms and complications of the disease, but in most cases there is no cure. (Some researchers believe that bone marrow transplantation may offer a cure in a small percentage of cases.) Over the past 30 years, doctors have learned a great deal about the disease. They know what causes it, how it affects the body, and how to treat many of the complications. Today, with good health care, many people with the disease:
Anemia
Anemia is the term for having a shortage of red blood cells in the blood. In sickle cell anemia, this shortage of red blood cells occurs because sickle cells do not last very long. Red blood cells are produced in the spongy marrow inside the large bones of the body. The bone marrow constantly makes new red blood cells to replace old ones. Normal red blood cells last about 120 days in the bloodstream and then die. Sickle cells die much faster, usually after only about 10 to 20 days. The bone marrow can't make new red blood cells fast enough to replace the dying ones, so anemia (low level of red blood cells) results.
Sickle cell trait versus anemia
The condition called sickle cell trait is different from sickle cell anemia. A person with sickle cell trait does not have the disease but carries the gene that causes the disease. People with sickle cell trait can pass the gene on when they have children.
Children who inherit only one copy of the sickle cell gene (from one parent) will not have sickle cell anemia. They will have sickle cell trait.
People with sickle cell trait:
When two people with sickle cell trait have a baby, there is a:
Who is at risk?
Sickle cell anemia affects millions of people throughout the world. It is common in people whose families come from any of these areas of the world:
In the United States, sickle cell anemia affects about 72,000 people. The families of most Americans who are affected come from Africa. In the United States, the disease occurs in about:
Signs and symptoms
The signs and symptoms of sickle cell anemia are different in each person. Some people have mild symptoms. Others have very severe symptoms and are often hospitalized for treatment.
The most common signs or symptoms are related to:
The general signs or symptoms of anemia are:
Pain is the symptom of sickle cell anemia that most people know. In both children and adults, pain may result from blocked flow of blood and oxygen. Painful events may occur in any body organ or joint. Some patients have these painful crises less than once a year. Others may have 15 or more crises in a year. The pain can be acute (sudden), chronic (long lasting), or a mixture of the two.
Other complications of sickle cell anemia include:
Diagnosis
Forty-four States, the District of Columbia, Puerto Rico, and the Virgin Islands currently test all newborns for sickle cell anemia. This testing is available by request in the other six States. The test can show if the newborn infant has sickle cell anemia or carries the sickle cell trait. The test uses blood from the same blood samples as other routine newborn screening tests. If the first test shows that the sickle-shaped hemoglobin is present, a second blood test is done to confirm the diagnosis.
Early diagnosis of sickle cell anemia is very important so that children who have the disease can get proper treatment.
It is also possible to identify sickle cell anemia before birth. This is done by getting a sample of amniotic fluid or tissue taken from the placenta. This test can be done as early as the first few months of pregnancy.
Treatment
Effective treatments exist for the symptoms and complications of sickle cell anemia, but in most cases there is no cure. Some researchers believe that bone marrow transplantation may offer a cure in a small percentage of cases. Researchers are working on developing new treatments for sickle cell anemia, including gene therapy and more safe and effective bone marrow transplants.
People with sickle cell anemia need regular medical care. Some doctors and clinics specialize in treating people with the disease.
The goals of treating sickle cell anemia are to relieve pain, prevent infections, and control complications if they occur. The treatments include:
Treating pain
Painful crises are the leading cause of emergency room visits and hospitalizations of people with sickle cell anemia. The usual treatments for acute pain crises are pain-killing medicines and fluids, either by mouth or through a vein, to prevent dehydration. The pain-killing medicines most often used are:
The treatment of patients with mild-to-moderate pain usually begins with NSAIDs or acetaminophen. If pain continues, an opioid may be added. Moderate-to-severe pain is treated with opioids. The opioid may be used alone or together with NSAIDs or acetaminophen.
In adult patients with severe sickle cell anemia, doctors may give a medicine called hydroxyurea to reduce the frequency of painful crises. This medicine is used only to prevent these crises, not to treat them when they occur. Given daily, it can reduce the frequency of painful crises and of acute chest syndrome. Patients taking the medicine may also need fewer blood transfusions. Patients taking hydroxyurea must be monitored carefully because the medicine can cause serious side effects, including an increased risk of dangerous infections. Because of the risks of the medicine, it is usually only used in adults with severe complications of sickle cell anemia. It is not approved for use in children.
Preventing infections
Infection is a major complication of sickle cell anemia. In fact, pneumonia is the leading cause of death in children with the disease. Other infections common in people with the disease include meningitis, influenza, and hepatitis.
To prevent infections in babies and young children, treatments include:
Children with sickle cell anemia should have a flu shot each year. If a child with sickle cell anemia shows early signs of an infection, such as fever, it is very important to get treatment right away.
Adults with sickle cell anemia should also have a flu shot every year. In addition, they should be vaccinated for pneumonia.
Preventing eye damage
Children may develop damage to the blood vessels in the back of their eyes. Parents should ask the child's doctor about regular checkups with an eye doctor who specializes in diseases of the retina. The retina is a thin layer of tissue inside the back of the eye.
Blood transfusions
Blood transfusions are used to treat worsening anemia and sickle cell complications. A sudden worsening of anemia resulting from infection or enlargement of the spleen is a common reason for a transfusion. Some, but not all, patients need transfusions to prevent life-threatening events such as stroke or pneumonia.
Treating complications
Acute chest syndrome is a frequent cause of death in children and adults with sickle cell anemia. Treatment usually requires hospitalization and may include oxygen therapy, transfusions, antibiotics, and pain medicine.
Hand-foot syndrome is an early complication seen in sickle cell anemia. The syndrome may start at less than 1 year of age. Treatment includes the use of pain medicine and fluids.
Leg ulcers may be treated with cleansing solutions and medicated creams or ointments. Skin grafts may be needed if the condition continues. Leg ulcers can be painful, and patients may be given strong pain medicine. Bed rest and keeping the leg raised to reduce swelling are useful, although not always possible.
Regular health care for children
Children with sickle cell anemia should get regular health care, just like children without the disease. They need to have their growth checked and to get the usual shots that all children receive.
Before age 2, children with sickle cell anemia should see the doctor every 2 or 3 months. After age 2, children should see the doctor at least every 6 months. These visits are good chances for parents to talk with their child's health care provider and ask questions about the child's care.
Penicillin is generally given to all children with sickle cell anemia until age 5. Many patients are prescribed a vitamin called folic acid (folate) to help prevent some of the complications of sickle cell anemia.
New treatments
Today, research on sickle cell anemia is looking at new medicines, bone marrow transplants, and gene therapy. The hope is that these studies can provide new treatments and find a possible cure for sickle cell anemia. Researchers are also looking for a way to predict the severity of the disease.
Bone marrow transplant can be a very effective treatment for sickle cell anemia, but, because of its risks, only some patients can or should have this procedure. The bone marrow transplant procedure carries the risk of serious complications and even death. It is usually reserved for younger patients with severe disease, but the decisions are made on a case-by-case basis. It requires a donation of bone marrow from a closely matched donor, usually a close family member, who does not have the disease.
Living with sickle cell anemia
If you have sickle cell anemia, it is important to take good care of yourself and see your doctor regularly. Things you need to do to take care of your health include:
Caring for a child with sickle cell anemia
If your child has sickle cell anemia, you should learn as much about the disease as possible. This will help you recognize early signs of problems, such as fever or chest pain, and seek early treatment. Sickle cell centers and clinics can provide information and counseling to help you handle the stresses of coping with this serious chronic disease.
Make sure that your child gets daily penicillin up to age 5 to prevent serious infections. Children should also have a flu shot every year and a vaccination against pneumonia. Keep a thermometer on hand, and know how to use it. Call a doctor if your child has a temperature above 101°F (38.5°C). Talk to your child's doctor about your child's treatment and the best ways to help keep your child as healthy as possible.
School-aged children should participate in physical education. Teachers should allow children with sickle cell anemia to rest if they tire and to drink fluids after exercise. Children and teenagers may also play competitive sports. Coaches should watch for signs of fatigue and allow the athlete to rest.
Education and job training
If you have sickle cell anemia, it is important that you complete your education. You should not seek jobs that require strenuous work, long work hours, or exposure to extreme temperatures.
Genetic counseling
People who are planning to have children should find out if they have sickle cell trait (which means they carry the sickle cell gene). If both parents have sickle cell trait, they should get genetic counseling. The counselor can tell what the chances are that their child will have sickle cell trait or sickle cell anemia. Accurate diagnostic tests and information are available from health departments, neighborhood health centers, medical centers, and clinics that care for people with sickle cell anemia.
With good health care, many people with sickle cell anemia are in reasonably good health much of the time and can live productive lives. People with sickle cell anemia are living longer today than in the past. Many patients with sickle cell anemia now live into their forties, fifties, and beyond.

